使用Python进行(生物)图像分析:使用Matplotlib读取和加载显微图像
Python进行图像分析:使用Matplotlib加载显微图像
在过去的二十年中,光学显微镜领域得到了显著的进展,得益于诸如共焦激光扫描显微镜(CLSM)、多光子激光扫描显微镜(MPLSM)、光片荧光显微镜(LSFM)和超分辨显微镜等开创性技术的引入[1]。这些技术极大地提高了捕捉图像的深度穿透性、生存能力和分辨率的能力。然而,在这些显著进展的同时,这些复杂技术的出现也带来了在处理大量生成的数据方面的巨大挑战。有效地可视化、分析和传播这些复杂数据集已成为充分利用这些前沿显微镜技术的重要方面。在本教程中,我们将探讨如何使用强大的Matplotlib Python库读取和可视化复杂的2D、3D、4D和5D光学显微镜图像。
目录
- 1.1 图像与像素
- 1.2 通道
- 1.3 4D图像(x、y、通道和Z切片)
- 1.4 投影
- 1.5 5D图像(x、y、通道、Z切片和时间)
- 1.6 结论
- 1.7 参考文献
# 导入库import numpy as npimport matplotlib.pyplot as pltimport skimage as skfrom matplotlib_scalebar.scalebar import ScaleBar图像与像素
显微镜图像是以数字方式记录的,为了实现这一点,形成最终图像的光子流必须被分割成小的几何子单元,即像素[2]。对计算机而言,每个像素只是一个数字(图1),当显示图像数据时,像素的值通常被转换为方块;这些方块只是一种有助于我们快速了解图像内容的可视化方式[3]。
![图1.真菌黄孢菌的PilA-GFP内质体蛋白[5]、[6]的亚细胞定位的显微图像,放大区域(红色插图)显示像素值。图像是使用Leica TCS SP8 MP多光子显微镜捕获的(作者提供)](https://miro.medium.com/v2/format:webp/1*L4Luyzm04ji5moFMT36iEw.png)
在显微镜和特别是荧光显微镜中,显微图像通常是在没有颜色信息的情况下获取的(使用单色相机或光电倍增器),并且(通常)使用单色伪彩色查找表(LUT)进行可视化。查找表(LUTs;有时也称为颜色映射)确定了灰度值如何转换为颜色值。为了可视化显微图像,我们通常使用灰度色调(灰度LUT),或者在我们希望增加读者对暗特征的视觉敏感度的情况下,我们反转图像,将暗值映射为亮值,反之亦然[4]。
所有发布的显微图像应包含一个标度尺,作为图像中观察到的结构或特征的大小的视觉参考。
- “大型语言模型真的需要那么多层吗?这项人工智能研究揭示了模型的效率:大型语言模型中必不可少的组件的探索”
- GPT4Readability — 再也不用写 README 了
- 一项新的AI研究介绍了REV:AI研究中的一项重大突破——一种新的信息论度量方法,用于评估自由文本理由中的新颖且与标签相关的信息
# 使用scikit-image读取图像img = sk.io.imread("Single_channel_eisosome.tif")# 打印图像的尺寸print(f"图像尺寸:{img.shape[0]} x {img.shape[1]} 像素")# 创建一个带有两个子图的图形fig, axs = plt.subplots(1, 2, figsize=(6, 4))# 在第一个子图中绘制灰度图像axs[0].imshow(img,cmap="gray")axs[0].axis('off')# 在子图中添加比例尺scalebar = ScaleBar(0.0721501, 'um', length_fraction=0.2, height_fraction=0.02, location='lower right')axs[0].add_artist(scalebar)axs[0].set_title("灰度图像")# 在第二个子图中绘制反转的灰度图像axs[1].imshow(img,cmap="gray_r")axs[1].axis('off')# 在子图中添加比例尺scalebar = ScaleBar(0.0721501, 'um', length_fraction=0.2, height_fraction=0.02, location='lower right')axs[1].add_artist(scalebar)axs[1].set_title("反转图像")# 调整子图之间的间距plt.tight_layout()图像尺寸:589 x 589 像素
通道
在显微镜下,不同的通道代表用于捕捉和可视化样本中特定组分或结构的不同光谱范围。每个通道对应于特定的波长范围或发射光谱,使研究人员能够同时对样本中的不同分子、结构或事件进行成像。为了优化视觉识别,建议在大多数荧光成像应用中使用单独的黑白通道。可以创建最终的合并彩色图像以显示叠加效果,确保对于常见的色盲读者也具有可访问性。在下面的代码中,我们可视化了A. nidulans的分生孢子中PilA-GFP(使用灰度查找表)和Histone H1-mRFP(使用灰度查找表)的亚细胞定位,以及它们的叠加图像(PilA显示为品红色,Histone H1显示为绿色)[7]。
# 使用scikit-image读取图像img = sk.io.imread("3_channel_eisosome.tif")# 打印图像的尺寸print(f"图像的尺寸为{img.shape[0]} x {img.shape[1]}像素,具有{img.shape[2]}个通道")# 分离通道h1_histone = img[:,:,0]pila = img[:,:,1]brightfield = img[:,:,2]# 创建一个包含四个子图的图形fig, axs = plt.subplots(1, 4, figsize=(12, 4))# 在第一个子图中绘制H1 histoneaxs[0].imshow(h1_histone,cmap="gray")axs[0].axis('off')axs[0].set_title("H1 histone (灰度图像)")# 在第二个子图中绘制PilA蛋白axs[1].imshow(pila,cmap="gray")axs[1].axis('off')axs[1].set_title("PilA蛋白(灰度图像)")# 在第三个子图中绘制亮场图像,并带有比例尺axs[2].imshow(brightfield,cmap="gray")axs[2].axis('off')scalebar = ScaleBar(0.0721501, 'um', length_fraction=0.2, height_fraction=0.02, location='lower right')axs[2].add_artist(scalebar)axs[2].set_title("亮场图像")# 将2D切片转换为3D数组h1_histone_3d = np.atleast_3d(img[:,:,0]) #将2D切片转换为3D数组pila_3d = np.atleast_3d(img[:,:,1]) #将2D切片转换为3D数组# 定义颜色color_h1 = np.asarray([1, 0, 1]).reshape((1, 1, -1)) #创建一个1x1x3的数组,品红色color_pila = np.asarray([0, 1, 0]).reshape((1, 1, -1)) #创建一个1x1x3的数组,绿色# 将图像中的每个像素乘以颜色值h1_magenta = h1_histone_3d * color_h1pila_green = pila_3d * color_pila# 合并彩色通道merged = np.clip(h1_magenta + pila_green,0,255)# 在第四个子图中绘制合并后的通道axs[3].imshow(merged)axs[3].axis('off')axs[3].set_title("合并通道")# 调整子图之间的间距plt.tight_layout()图像的尺寸为589 x 589像素,具有3个通道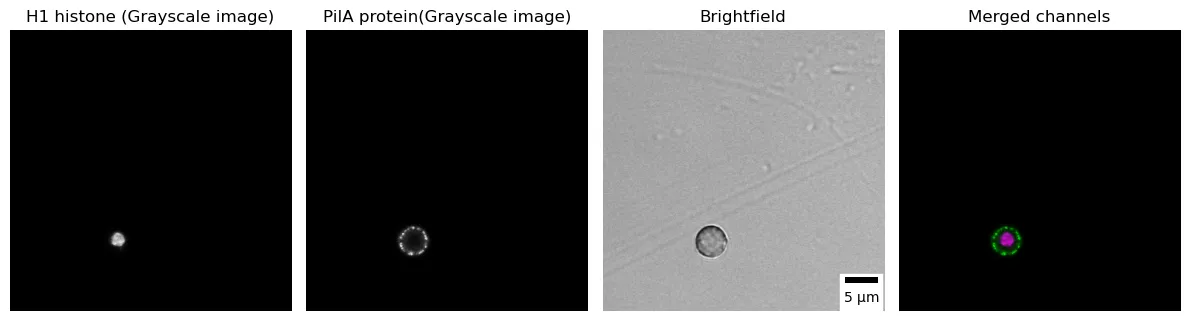
4D图像(x,y,通道和Z切片)
在共聚焦显微镜中,我们经常在Z轴上沿着不同焦平面捕捉图像的“切片”(Z堆叠)。然后将这些切片堆叠在一起,重建成3D图像。Z堆叠为我们提供了有关样本内结构的空间分布、形态和关系的宝贵见解。在下面的代码中,我们通过在Z轴上捕捉四个“切片”(实际上,下面的图像代表一个超堆栈,即由x、y、z和通道组成的4D组合)来展示A. nidulans的分生孢子中PilA-GFP和Histone H1-mRFP的亚细胞定位。这种方法使我们能够研究这些蛋白在三维空间中的分布和关系。
# 使用scikit-image读取图像
img = sk.io.imread("3_channel_zstack_eisosome.tif")
# 打印图像的尺寸
print(f"图像的尺寸为 {img.shape[1]} x {img.shape[2]} 像素,通道数为 {img.shape[3]},切片数为 {img.shape[0]}")
# 分离通道
h1_histone = img[:,:,:,0]
pila = img[:,:,:,1]
brightfield = img[:,:,:,2]
# 创建带有子图的图形
fig, axs = plt.subplots(3, 4, figsize=(12, 7))
# 绘制H1 histone通道的每个切片
for i in range(img.shape[0]):
axs[0][i].imshow(h1_histone[i,:,:],cmap="gray")
axs[0][i].axis('off')
axs[0][i].set_title(f"(Z = {i+1}) H1 histone")
scalebar = ScaleBar(0.0721501, 'um', length_fraction=0.2, height_fraction=0.02, location='lower right')
axs[0, -1].add_artist(scalebar)
# 绘制PilA protein通道的每个切片
for i in range(img.shape[0]):
axs[1][i].imshow(pila[i,:,:],cmap="gray")
axs[1][i].axis('off')
axs[1][i].set_title(f"(Z = {i+1}) PilA protein")
scalebar = ScaleBar(0.0721501, 'um', length_fraction=0.2, height_fraction=0.02, location='lower right')
axs[1, -1].add_artist(scalebar)
# 创建H1 histone和PilA protein通道的合并图像
for i in range(img.shape[0]):
h1_histone_3d = np.atleast_3d(h1_histone[i,:,:]) #将2D切片转换为3D数组
pila_3d = np.atleast_3d(pila[i,:,:]) #将2D切片转换为3D数组
color_h1 = np.asarray([1, 0, 1]).reshape((1, 1, -1)) #创建一个1x1x3的数组,表示品红色
color_pila = np.asarray([0, 1, 0]).reshape((1, 1, -1)) #创建一个1x1x3的数组,表示绿色
h1_magenta = h1_histone_3d * color_h1
pila_green = pila_3d * color_pila
merged = np.clip(h1_magenta + pila_green,0,255)
axs[2][i].imshow(merged)
axs[2][i].axis('off')
axs[2][i].set_title(rf"(Z = {i+1}) H1和PilA")
scalebar = ScaleBar(0.0721501, 'um', length_fraction=0.2, height_fraction=0.02, location='lower right')
axs[2, -1].add_artist(scalebar)
# 调整子图之间的间距
plt.tight_layout()
图像的尺寸为589 x 589像素,通道数为3,切片数为4
投影
一种可视化Z-stack的方法是逐个研究每个切片,就像上面的代码演示的那样。然而,当处理多个切片时,这种方法变得繁琐。一种高效的方法是通过计算z-projection来总结Z-stack中的信息[3]。通过进行z-projection,我们可以根据我们要回答的特定问题生成各种类型的投影。例如,我们可以获得最大、最小、平均或标准差投影。在最大投影中,我们创建一个2D图像来表示最大强度,捕获最亮的特征,沿着特定的轴,通常是z轴。类似地,在平均、最小和标准差投影中,我们生成代表强度的平均值、最小值或扩展的2D图像,沿着特定的轴。
# 使用scikit-image读取图像
img = sk.io.imread("3_channel_zstack_eisosome.tif")
# 分离通道
h1_histone = img[:,:,:,0]
pila = img[:,:,:,1]
brightfield = img[:,:,:,2]
# 创建带有子图的图形
fig, axs = plt.subplots(1, 4, figsize=(12, 7))
# 要显示的投影名称列表
projections = ['最小投影', '平均投影', '最大投影', '标准差投影']
# 要计算的投影名称列表
np_projection = [np.min, np.mean, np.max, np.std]
# 计算投影;保留相同的dtype输出
for i in range(len(projections)):
h1_histone_3d = np.atleast_3d(np_projection[i](h1_histone, axis=0).astype(h1_histone.dtype)) #将2D切片转换为3D数组
pila_3d = np.atleast_3d(np_projection[i](pila, axis=0).astype(pila.dtype)) #将2D切片转换为3D数组
color_h1 = np.asarray([1, 0, 1]).reshape((1, 1, -1)) #创建一个1x1x3的数组,表示品红色
color_pila = np.asarray([0, 1, 0]).reshape((1, 1, -1)) #创建一个1x1x3的数组,表示绿色
h1_magenta = h1_histone_3d * color_h1
pila_green = pila_3d * color_pila
merged = np.clip(h1_magenta + pila_green,0,255)
axs[i].imshow(merged)
scalebar = ScaleBar(0.0721501, 'um', length_fraction=0.2, height_fraction=0.02, location='lower right')
axs[i].add_artist(scalebar)
axs[i].axis('off')
axs[i].set_title(projections[i])
5D图像(x,y,通道,Z切片和时间)
共聚焦5D成像是生物学中一种有价值的工具,可以提供有关细胞和亚细胞水平上动态过程的重要见解。这些图像包括x、y、z切片和通道以及时间,使得可以对时间变化进行可视化和分析。在以下代码中,我们通过在每个时间点上呈现PilA-GFP和Histone H1-mRFP的最大投影(合并图像),展示了共聚焦5D成像的重要性,每个时间点代表大约20分钟。
# 使用scikit-image读取5D图像堆栈img = sk.io.imread("3_channel_zstack_time_eisosome.tif")print(f"数据集具有{img.shape[2]} x {img.shape[3]}像素,具有{img.shape[4]}个通道,{img.shape[1]}个切片和{img.shape[0]}个时间点")# 分离通道h1_histone = img[:,:,:,:,0]pila = img[:,:,:,:,1]brightfield = img[:,:,:,:,2]# 创建带有子图的图像fig,axs = plt.subplots(6, 7, figsize=(19,14))# 计算投影(在输出中保留相同的数据类型)并为每个时间点创建合并图像time =0for i in range (img.shape[0]): # 计算H1 histone通道的最大投影 h1_histone_3d = np.atleast_3d(np.max(h1_histone[i,:,:,:], axis=0).astype(h1_histone.dtype)) # 计算PilA通道的最大投影 pila_3d = np.atleast_3d(np.max(pila[i,:,:,:], axis=0).astype(pila.dtype)) # 计算brightfield通道的最小投影 brightfield_3d = np.atleast_3d(np.min(brightfield[i,:,:,:], axis=0).astype(brightfield.dtype)) # 为每个通道定义颜色 color_h1 = np.asarray([1, 0, 1]).reshape((1, 1, -1)) #创建一个1x1x3的数组 - 品红色 color_pila = np.asarray([0, 1, 0]).reshape((1, 1, -1)) #创建一个1x1x3的数组 - 绿色 color_brightfield = np.asarray([0.5, 0.5, 0.5]).reshape((1, 1, -1)) #创建一个1x1x3的数组 - 灰色 # 通过组合通道创建合并图像 h1_magenta = h1_histone_3d * color_h1 pila_green = pila_3d * color_pila brightfield_gray = brightfield_3d* color_brightfield # merged = np.clip(h1_magenta + pila_green+brightfield_gray,0,255) merged = np.clip(h1_magenta + pila_green,0,255)# 在相应的子图中绘制合并图像 axs[i // 7, i % 7].imshow(merged) axs[i // 7, i % 7].axis('off') axs[i // 7, i % 7].set_title(f"{time} 分钟") scalebar = ScaleBar(0.0721501, 'um', length_fraction=0.2, height_fraction=0.02, location='右下角') axs[5, 1].add_artist(scalebar) time +=20plt.tight_layout()plt.show()数据集具有589 x 589像素,具有3个通道,4个切片和37个时间点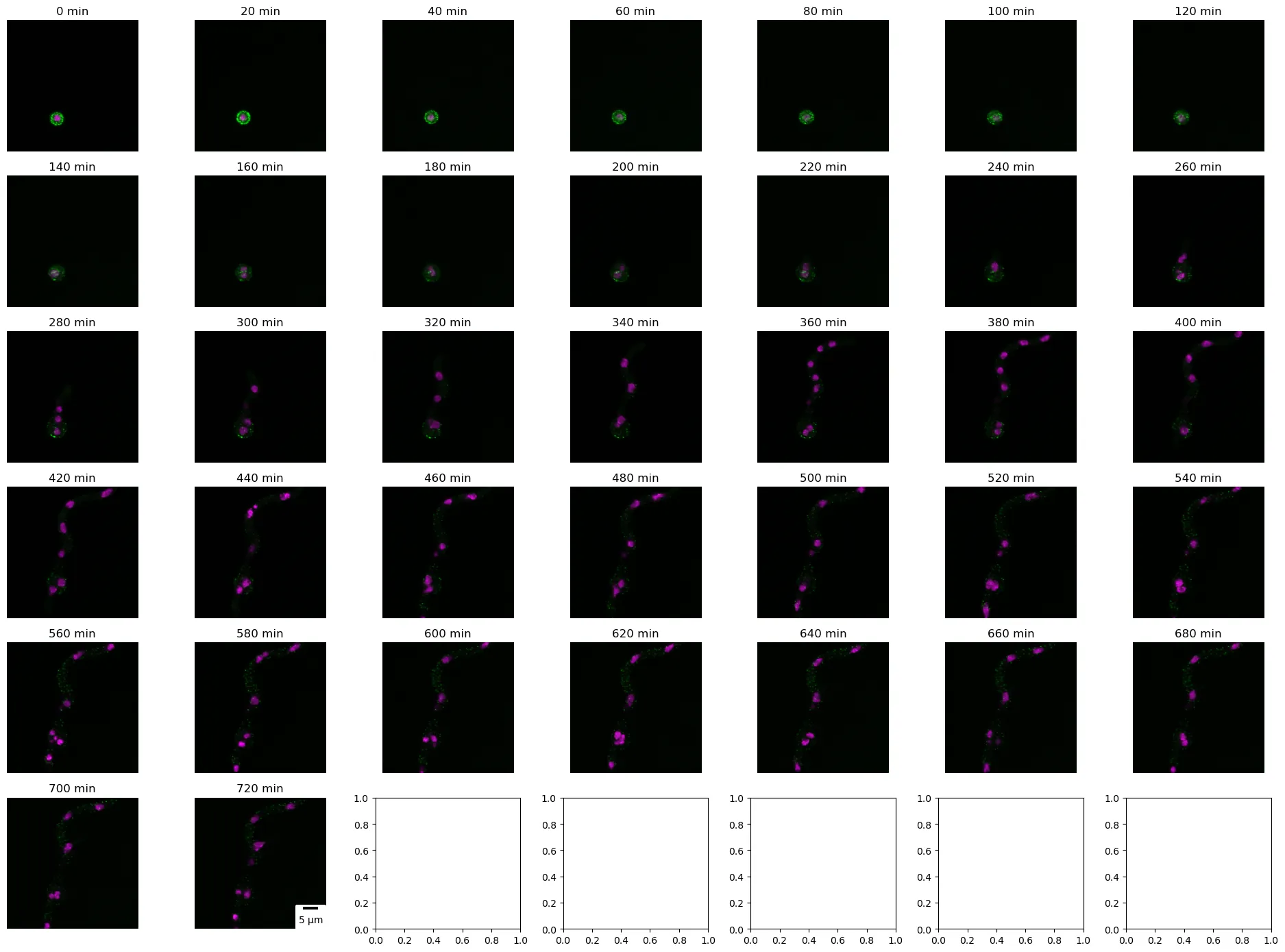
在不同的时间点上显示真菌A. nidulans的亮场切片(如下所示)。
# 使用scikit-image读取5D图像堆栈img = sk.io.imread("3_channel_zstack_time_eisosome.tif")# 分离通道h1_histone = img[:,:,:,:,0]pila = img[:,:,:,:,1]brightfield = img[:,:,:,:,2]# 创建带有子图的图像fig,axs = plt.subplots(6, 7, figsize=(19,14))# 计算投影(在输出中保留相同的数据类型)并为每个时间点创建合并图像time =0for i in range (img.shape[0]):# 选择Brihgtfield通道的切片 brightfield_2_slice = brightfield[i,2,:,:]# 在相应的子图中绘制第二个切片 axs[i // 7, i % 7].imshow(brightfield_2_slice, cmap="gray") axs[i // 7, i % 7].axis('off') axs[i // 7, i % 7].set_title(f"{time} 分钟") scalebar = ScaleBar(0.0721501, 'um', length_fraction=0.2, height_fraction=0.02, location='右下角') axs[5, 1].add_artist(scalebar) time +=20plt.tight_layout()plt.show()

结论
在本教程中,我们将深入了解阅读和可视化复杂的光学显微图像的过程,包括2D、3D、4D和5D。虽然matplotlib是一个强大的数据可视化库,但它可能不是最适合用于可视化3D、4D和5D图像(如显微图像)的选择。虽然matplotlib可以有效处理2D图像,但是可视化高维图像可能具有挑战性和限制。然而,有解决方案:
- Microfilm [9],[10],简化了多通道图像的绘制。
- 还有Napari [8],一个专门用于可视化和探索多维科学数据(包括显微图像)的流行Python库。Napari提供了一个交互式和多功能的界面,超出了matplotlib的能力。
在我们即将发布的教程中,我们将深入研究Napari并详细了解其可视化高维图像的功能。
我已经准备了一个Jupyter Notebook来配合本博文,可以在我的GitHub上查看。

参考文献:
[1] C. T. Rueden和K. W. Eliceiri,“多维生物图像数据的可视化方法”,BioTechniques杂志,第43卷,第1S期,第S31-S36页,2007年7月,doi:10.2144/000112511。
[2] J. B. Pawley,“点、像素和灰度级别:数字化图像数据”,收录于《生物共焦显微镜手册》中,J. B. Pawley主编,波士顿,马萨诸塞州:Springer US,2006年,第59-79页,doi:10.1007/978-0-387-45524-2_4。
[3] P. Bankhead,“生物图像分析简介-生物图像分析简介。” https://bioimagebook.github.io/index.html (访问日期:2023年6月29日)。
[4] J. Johnson,“看不见就不相信:改善您的荧光图像的可见性”,Mol. Biol. Cell杂志,第23卷,第5期,第754-757页,2012年3月,doi:10.1091/mbc.e11-09-0824。
[5] I. Vangelatos,K. Roumelioti,C. Gournas,T. Suarez,C. Scazzocchio和V. Sophianopoulou,“Aspergillus nidulans中Eisosome的组织”,Eukaryot. Cell杂志,第9卷,第10期,第1441-1454页,2010年10月,doi:10.1128/EC.00087-10。
[6] A. Athanasopoulos,C. Gournas,S. Amillis和V. Sophianopoulou,“AnNce102的特性及其在Eisosome稳定性和鞘脂生物合成中的作用”,Sci. Rep.杂志,第5卷,第1期,2015年12月,doi:10.1038/srep15200。
[7] A. P. Mela 和 M. Momany,“Aspergillus nidulans 细胞区域内组蛋白 H1 的核间扩散”,PloS One,vol. 13,no. 8,p. e0201828,2018,doi: 10.1371/journal.pone.0201828。
[8] “napari:Python 中的快速交互式多维图像查看器 — napari。”https://napari.org/stable/(访问日期:2023 年 7 月 4 日)。
[9] G. Witz,“microfilm。”2023 年 6 月 27 日。访问日期:2023 年 7 月 14 日。[在线]。可用:https://github.com/guiwitz/microfilm
[10] “Bio-image Analysis Notebooks — 生物图像分析笔记本。”https://haesleinhuepf.github.io/BioImageAnalysisNotebooks/intro.html(访问日期:2023 年 7 月 14 日)。






