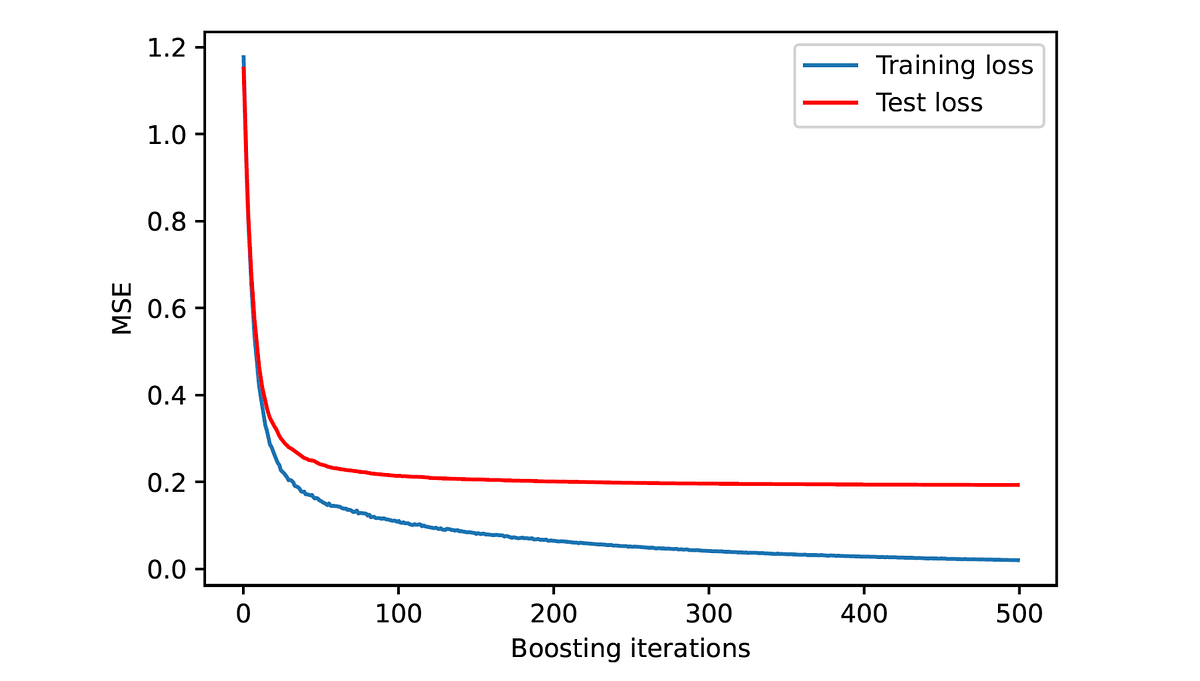
从位到生物学 #1:在计算生物学中使用LCS算法进行全局序列对准
计算生物学中使用LCS算法进行全局序列对准
对最长公共子序列问题和序列对齐问题进行逐步分析,解释它们的解决方案并突出它们的相似性和差异
介绍
欢迎!您找到了我在《从位到生物》系列中的第一篇文章,在这里,我探索了计算机科学课程中可能遇到的一般算法与计算生物学/生物信息学之间的联系,以使它们对没有正式生物学培训的人更加直观和易于理解。我花费了一些时间才确信您不一定需要正式的生物学背景才能在计算生物学(一个非常有趣的领域)中做出有意义的贡献,我希望我能说服您如果您处于类似情况的话,不妨试试:)
在本文中,我们将研究序列对齐,这是计算生物学中最基本的问题之一。DNA、RNA和蛋白质序列的对齐具有多种深远的影响,包括理解进化关系、进展功能注释、识别突变以及推动药物设计和精准医学。
目录
- 最长公共子序列(LCS)- LCS概述- LCS动态规划解决方案- LCS Python实现
- 全局序列对齐(GSA)- GSA概述- 评分系统- GSA动态规划解决方案- GSA Python实现
- LCS vs. 全局序列对齐:相似性和差异- 总结表
最长公共子序列(LCS)问题
最长公共子序列(LCS)是一个经典的计算机科学问题,它识别出2个或多个输入序列之间的所有有效子序列中最长的子序列。
首先,让我们通过注意子序列和子字符串之间的一个重要区别来澄清LCS问题的陈述。以序列"ABCDE"为例:
- 子字符串由原始顺序中连续的字符组成。例如,有效的子字符串包括
"ABC"、“CD”等,但不包括“ABDE”或“CBA”。 - 子序列则不必连续,但必须按照原始顺序。例如,有效的子序列包括
“AC”和“BFG”。
因此,LCS是2个或多个序列之间所有可能子序列中的最长子序列。下面是一个快速示例:

实际应用中,LCS在涉及更大的文本数据时具有许多应用,例如抄袭检测¹或Unix的diff命令,这需要一种高效且可靠、可扩展的算法解决方案。
形式化LCS解决方案
下面是LCS的三步动态规划解决方案的梳理:
- 初始化(创建一个m×n矩阵,其中m和n分别是序列1和序列2的长度)。
- 根据下面给出的递推公式填充矩阵。
- 回溯以找到LCS及其长度。
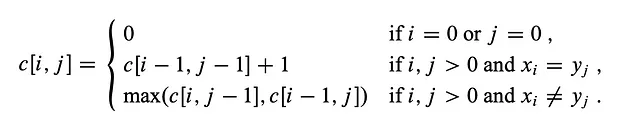
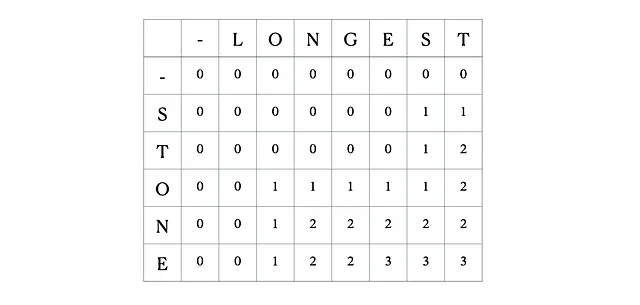
考虑输入的“LONGEST”和“STONE”。根据上述的递归关系,我们填充DP矩阵,然后进行回溯,如下所示:
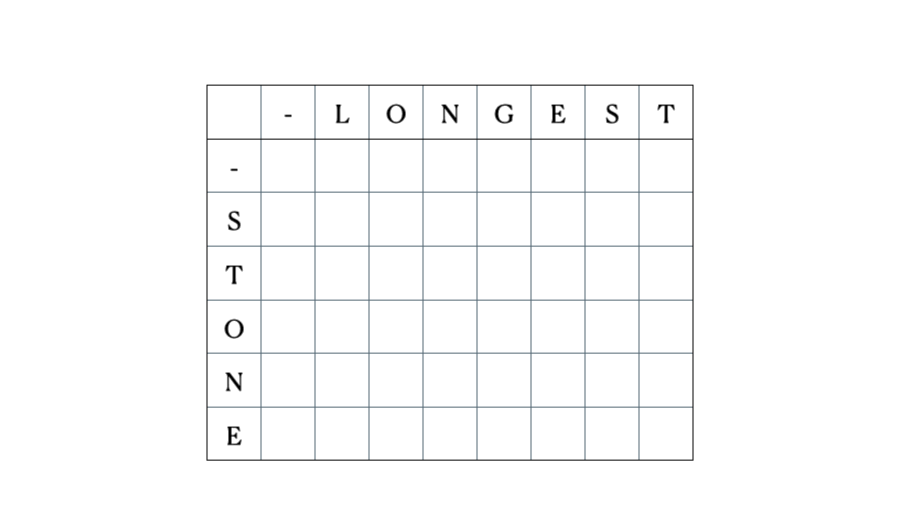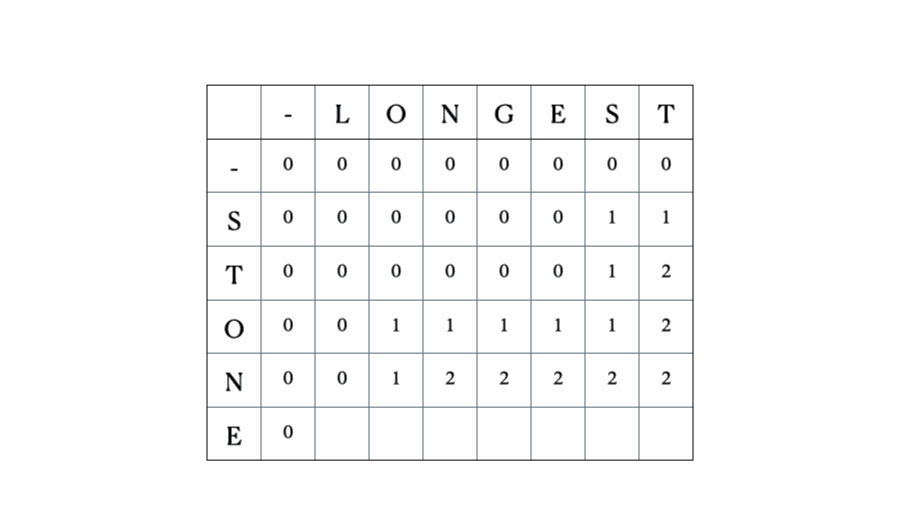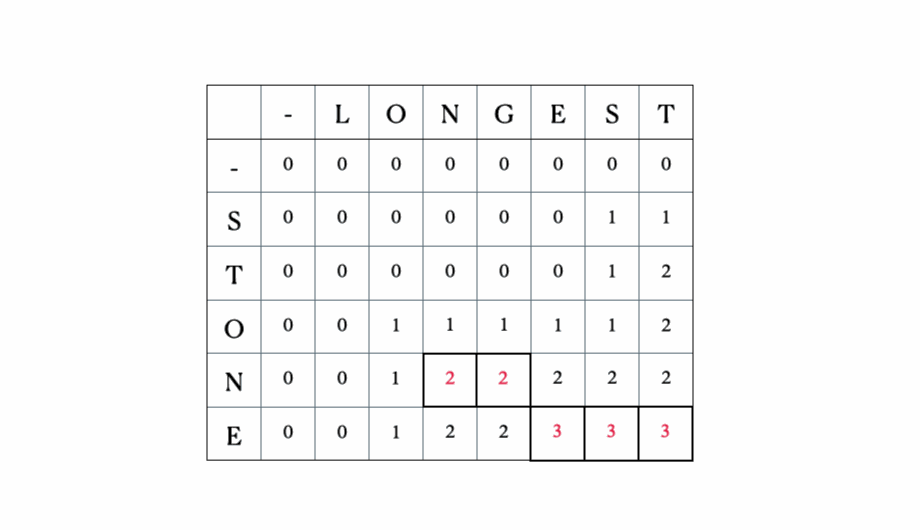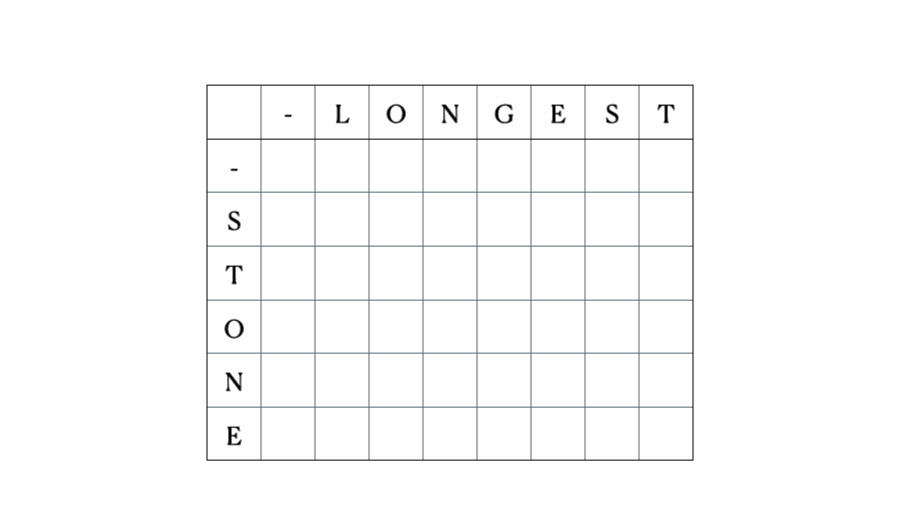
这是完整的DP表:
这可以转化为以下Python实现:
# 最长公共子序列
def lcs(seq1, seq2):
m = len(seq1)
n = len(seq2)
# 构建dp表
table = [[0 for x in range(n+1)] for x in range(m+1)]
# 基于CLRS中的递归公式(15.9)填充表格
for i in range(m+1):
for j in range(n+1):
if i == 0 or j == 0: # 情况1
table[i][j] = 0
elif seq1[i-1] == seq2[j-1]: # 情况2
table[i][j] = table[i-1][j-1] + 1
else: # 情况3
table[i][j] = max(table[i-1][j], table[i][j-1])
# 从右下角的单元格开始回溯
index = table[m][n]
lcs = [""] * (index+1)
lcs[index] = ""
i = m
j = n
while i > 0 and j > 0:
if seq1[i-1] == seq2[j-1]: # 将匹配的字符添加到LCS中
lcs[index-1] = seq1[i-1]
i -= 1
j -= 1
index -= 1
elif table[i-1][j] > table[i][j-1]:
i -= 1
else:
j -= 1
# 打印最长公共子序列及其长度
print("序列1:" + seq1 + "\n序列2:" + seq2)
print("LCS:" + lcs + ",长度:" + str(len(lcs)))
示例用法:
seq1 = 'STONE'
seq2 = 'LONGEST'
lcs(seq1, seq2)
输出:
序列1:STONE
序列2:LONGEST
LCS:ONE,长度:3
现在,让我们来看一下这个算法在…中的应用 ↓
(全局) 序列比对问题
为什么我们要对DNA/RNA/蛋白质序列进行比对?
从直观上讲,对齐两个序列是一项任务,其目的是在两个序列之间定位相同的片段,从而评估它们之间的相似程度。从概念上来说相对简单,但在实践中却非常复杂,因为DNA和蛋白质序列在进化过程中可能发生无数变化。具体而言,这些变化的常见示例包括:
- 通过插入、删除或替换进行的点突变示例:- 插入示例:AAA → ACAA- 删除示例:AAA → AA- 替换示例:AAA → AGA
- 来自不同基因的序列或片段的融合
这些意外的变化可能会引入模糊和差异,掩盖了我们对给定序列的理解。这就是我们使用比对技术来评估序列之间的相似性并推断同源性等特征的原因。
我们如何对齐序列?
由于这些进化变化以及生物序列比较的特性,与LCS不同,我们不能只是丢弃“无法匹配”的元素并仅保留公共片段,在全局序列比对中,我们必须对齐所有字符,这意味着比对通常涉及3个组成部分:
- 匹配,用连字符
|表示匹配的碱基之间 - 不匹配,用空格表示不匹配的碱基之间
- 缺失插入,用破折号
—表示
这里有一个例子。假设我们有两个DNA序列“ACCCTG”和“ACCTGC”。有许多种方法可以将它们逐个碱基地进行匹配,考虑到我们之前讨论过的3种操作:
我们可以在没有任何缺失插入的情况下将它们对齐:
ACCCTGT||| |ACCTGCT这个对齐有0个缺失插入,3个匹配,然后是3个不匹配,最后是1个匹配。
或者我们可以允许缺失插入以换取更多的匹配:
ACCCTG-T||| || |ACC-TGCT这个对齐有6个总的匹配,2个缺失插入,0个不匹配(我们不将碱基-缺失对视为不匹配,因为缺失插入已经带有缺失惩罚)。
或者,如果我们真的愿意,我们也可以这样对齐,这样只有1个总匹配:
-ACC-CT-GT | ACCTG-CT--在任何一对序列之间,几乎总是可以生成技术上正确的对齐方式,就像我们刚才看到的那样。其中一些明显比其他的差,但也会有一些“大致相等的好”选项,比如上面的选项1和2。
那么,我们如何评估每个对齐的质量,选择最优对齐,并在需要时解决并列情况呢?
评分方案
1. ACCCTGT 2. ACCCTG-T 3. -ACC-CTGT ||| | ||| || | | ACCTGCT ACC-TGCT ACCTG-CT-在上面的例子中,每个对齐涉及匹配、不匹配和缺失插入的不同组合。
与LCS问题不同,其中一个子序列仅通过其长度来评估 —— 越长越好 —— 在序列对齐中,我们通常通过定义一个定量评分系统来衡量对齐的质量,该系统涵盖了每个碱基对之间的3个关键组成部分:
- 匹配奖励:当我们的序列中存在碱基对匹配时,将该分数添加到整体对齐分数中。
- 缺失惩罚:每次引入缺失时,从整体对齐分数中减去的惩罚值。
- 不匹配惩罚:每次发生不匹配时,从整体对齐分数中减去的惩罚值。
我们如何将此形式化为算法?
算法解决方案在结构上与LCS解决方案有很大的重叠。它也是一个3步骤的过程,称为Needleman-Wunsch算法:
- 初始化(创建一个 m × n 的矩阵,其中 m 和 n 是序列 1 和序列 2 的长度)
- 根据下面给出的递归公式填充矩阵
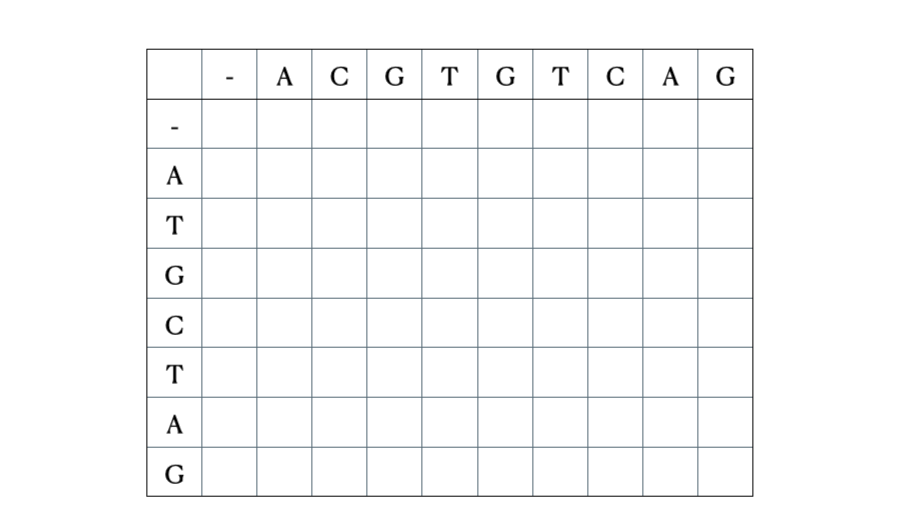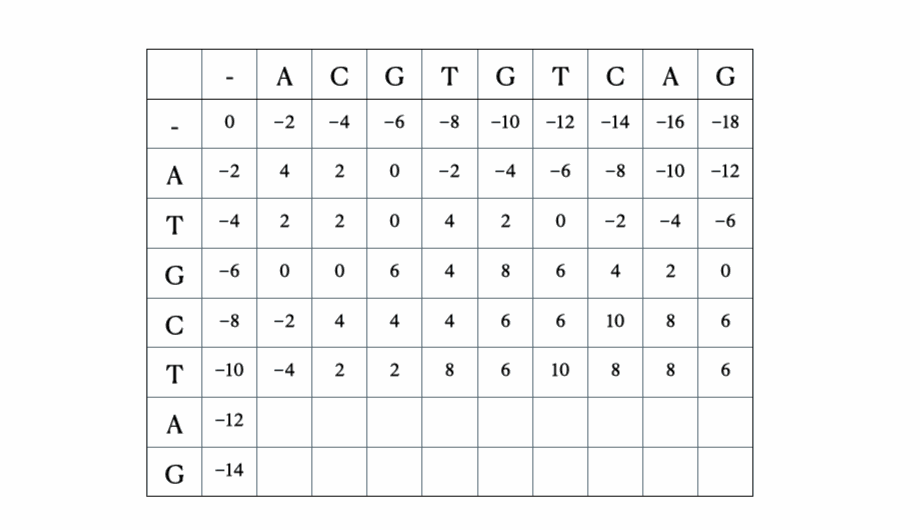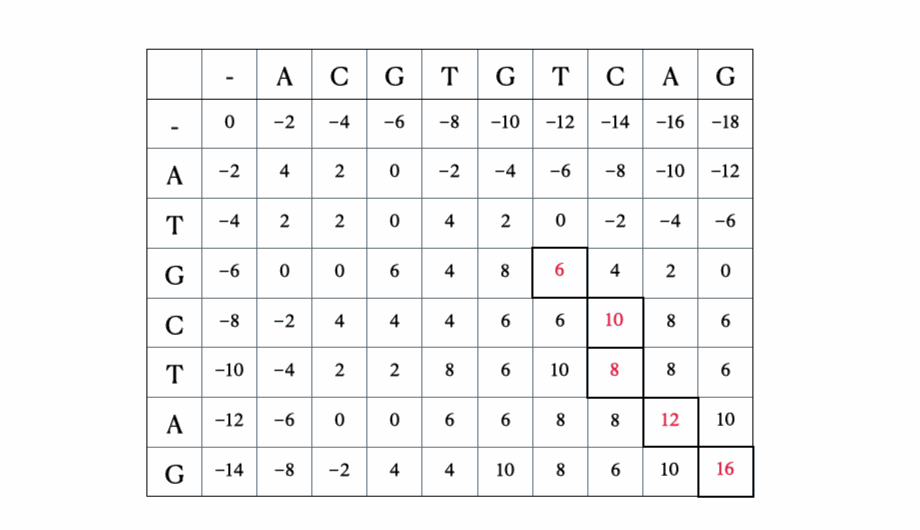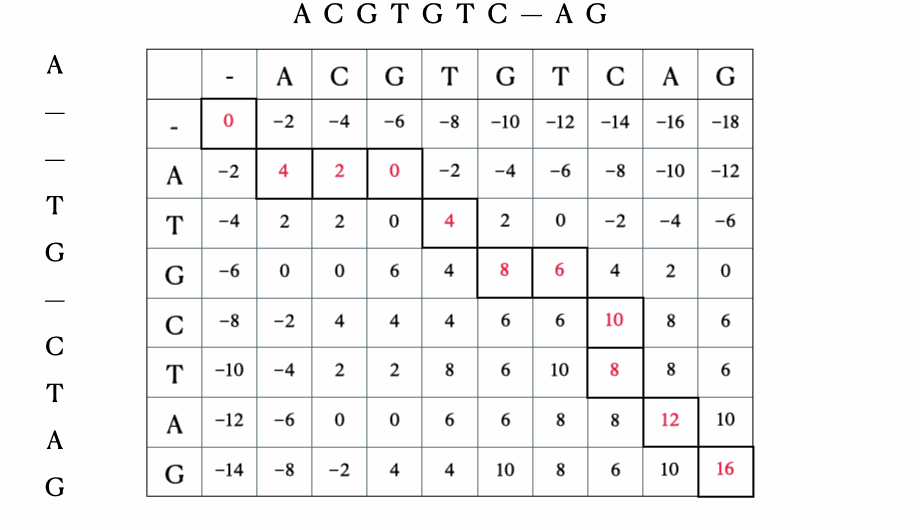
- 回溯以找到最优对齐以及其长度
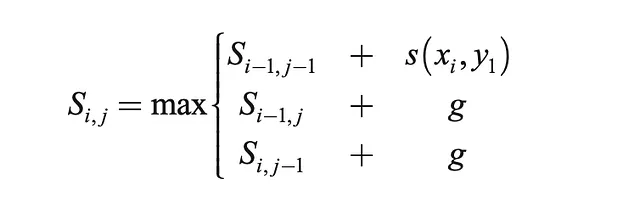
在我们进入解决方案的数学和Python形式之前,这里有一个使用序列“ACGTGTCAG”和“ATGCTAG”进行的快速示例:
Python解决方案
让我们使用一个名为GlobalSeqAlign的类来实现Python解决方案,该类负责填充矩阵以及回溯过程。
首先,我们构造一个包含5个关键信息的对象:序列1、序列2、匹配得分、不匹配惩罚和间隙惩罚。
class globalSeqAlign: def __init__(self, s1, s2, M, m, g): self.s1 = s1 self.s2 = s2 self.M = M self.m = m self.g = g然后,我们定义一个帮助函数,根据两个碱基之间是否匹配、不匹配或者是碱基间隙对来获取分数:
def base_pair_score(self, char1, char2): if char1 == char2: return self.M elif char1 == '-' or char2 == '-': return self.g else: return self.m现在,我们定义一个与之前看到的LCS函数非常相似的函数,它使用一个m × n的矩阵,根据递归公式填充矩阵,然后进行回溯以返回最大化总得分的最优对齐:
def optimal_alignment(self, s1, s2): m = len(s1) n = len(s2) score = [[0 for x in range(n+1)] for x in range(m+1)] # 计算得分表 for i in range(m+1): # 初始化为0 score[i][0] = self.g * i for j in range(n+1): # 初始化为0 score[0][j] = self.g * j for i in range(1, m+1): # 根据上述递归公式填充剩余单元格 for j in range(1, n+1): match = score[i-1][j-1] + self.base_pair_score(s1[j-1], s2[i-1]) delete = score[i-1][j] + self.g insert = score[i][j-1] + self.g score[i][j] = max(match, delete, insert) # 回溯,从右下角单元格开始 align1 = "" align2 = "" i = m j = n while i > 0 and j > 0: score_curr = score[i][j] score_diag = score[i-1][j-1] score_up = score[i][j-1] score_left = score[i-1][j] if score_curr == score_diag + self.base_pair_score(s1[j-1], s2[i-1]): # 匹配 align1 += s1[j-1] align2 += s2[i-1] i -= 1 j -= 1 elif score_curr == score_up + self.g: # 向上 align1 += s1[j-1] align2 += '-' j -= 1 elif score_curr == score_left + self.g: # 向左 align1 += '-' align2 += s2[i-1] i -= 1 while j > 0: align1 += s1[j-1] align2 += '-' j -= 1 while i > 0: align1 += '-' align2 += s2[i-1] i -= 1 return(align1[::-1], align2[::-1]) # 反转顺序一旦我们获得了最优对齐,我们可以继续进行分析,量化序列匹配的好坏。
量化相似性:百分比相似度
衡量“相似程度”的最简单方法之一是通过百分比相似度。我们将最优对齐的长度除以匹配区域的总长度。
例如:
序列1:ACACAGTCAT |||| |||||序列2:ACACTGTCAT共有10对中的9对匹配,得到90%的相似度。还有许多其他有趣的方法来分析对齐的质量,但序列对齐的基本原理是相同的。
LCS和全局序列对比
现在我们已经详细讨论了问题和解决方案,让我们将最长公共子序列(LCS)和全局序列对齐放在一起进行比较。
相似之处
- 目标:这两个问题都是在输入序列中寻找和最大化相似性和共同元素。
- 算法方法:这两个问题都可以使用动态规划来解决,其中包括一个DP矩阵/表,填写矩阵,并回溯以生成序列/对齐。
- 评分方案:这两个问题在序列匹配中都有与匹配、不匹配(和间隙)相关联的分数。这些分数在算法中被明确和隐含地用于检索最优子序列/对齐。
不同之处
- 评分系统:最长公共子序列(LCS)自动采用匹配得分=1,不匹配/间隙=0的评分方案;全局序列对齐有一个更复杂的评分方案,其中匹配得分=A,不匹配惩罚得分=B,间隙惩罚得分=C。
- 间隙处理:LCS通常将间隙视为与不匹配相等,因为根据子序列的定义,我们只保留匹配项;全局序列对齐通常给间隙指定惩罚分数,与不匹配惩罚分数不同,如果对不匹配/间隙有偏好的话。
- 对齐长度:LCS关注的是找到所有输入中的一个公共片段,忽略不属于该片段的元素;全局序列对齐匹配整个给定的序列,即使这意味着在输入序列长度不同时插入间隙。
下面是上述要点的摘要表:

结论
感谢您一直陪伴至此😼!在本文中,我们重点讨论了一个特定的动态规划问题——最长公共子序列,并与其在计算生物学中的应用进行了对比。
在本系列的后续文章中,我们还将研究其他主题,如隐马尔可夫模型、高斯混合模型和分类算法,以及它们如何解决计算生物学中的特定问题。再见,祝您编程愉快✌🏼!
[1] Baba, K., Nakatoh, T., & Minami, T. (2017). Plagiarism detection using document similarity based on distributed representation. Procedia Computer Science, 111, 382–387. https://doi.org/10.1016/j.procs.2017.06.038
[2] Cormen, T. H. (Ed.). (2009). Introduction to algorithms (3rd ed). MIT Press.
[3] Zvelebil, M. J., & Baum, J. O. (2008). Understanding bioinformatics. Garland Science.